Command line interface¶
$ limix -q download http://rest.s3for.me/limix/plink_example.tar.gz &\
limix -q download http://rest.s3for.me/limix/small_example.hdf5 &\
limix -q download http://rest.s3for.me/limix/small_example.csv.bz2 &\
limix -q download http://rest.s3for.me/limix/ex0/phenotype.gemma
$ limix -q extract plink_example.tar.gz &\
limix -q extract small_example.csv.bz2
Introduction¶
Limix now provides a couple of its functionalities via command line.
$ limix --help
Usage: limix [OPTIONS] COMMAND [ARGS]...
Options:
-v, --verbose / -q, --quiet Enable or disable verbose mode.
--version Show the version and exit.
-h, --help Show this message and exit.
Commands:
download Download file from the specified URL.
estimate-kinship Estimate a kinship matrix.
extract Extract a file.
remove Remove a file.
scan Perform genome-wide association scan.
see Show an overview of multiple file types.
You can quickly explore common file types used in genetics, as examples given bellow will demonstrate.
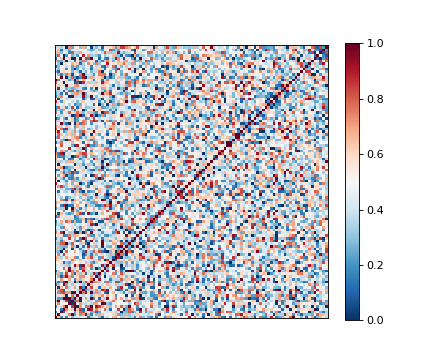
Kinship¶
Heatmap representing a plink kinship matrix. Setup:
limix download http://rest.s3for.me/limix/small_example.grm.raw.bz2
limix extract small_example.grm.raw.bz2
Command:
limix see small_example.grm.raw
(Source code, png)
Plink BED format¶
A preview of Plink files in BED format can be done via
$ limix see plink_example
Mapping files: 0%| | 0/3 [00:00<?, ?it/s]
Mapping files: 100%|██████████| 3/3 [00:00<00:00, 99.45it/s]
--------------------------------- Samples ---------------------------------
chrom snp cm pos a0 a1 i
0 22 snp_22_18958209 0.0 18958209 A G 0
1 22 snp_22_19597806 0.0 19597806 T C 1
2 22 snp_22_20171368 0.0 20171368 T C 2
3 22 snp_22_20179046 0.0 20179046 T C 3
4 22 snp_22_20828867 0.0 20828867 T C 4
5 22 snp_22_21350645 0.0 21350645 T C 5
6 22 snp_22_21387385 0.0 21387385 A T 6
7 22 snp_22_22061099 0.0 22061099 A G 7
8 22 snp_22_22329747 0.0 22329747 T G 8
9 22 snp_22_22800690 0.0 22800690 A T 9
10 22 snp_22_23106822 0.0 23106822 T C 10
11 22 snp_22_23705439 0.0 23705439 C T 11
12 22 snp_22_23805130 0.0 23805130 C A 12
13 22 snp_22_24677829 0.0 24677829 C T 13
14 22 snp_22_24944782 0.0 24944782 A G 14
15 22 snp_22_25825092 0.0 25825092 A G 15
16 22 snp_22_26247607 0.0 26247607 T C 16
17 22 snp_22_26585094 0.0 26585094 A T 17
18 22 snp_22_26675434 0.0 26675434 A C 18
19 22 indel:1I_22_27387365 0.0 27387365 TA T 19
20 22 snp_22_27520325 0.0 27520325 A T 20
21 22 snp_22_28178514 0.0 28178514 T C 21
22 22 snp_22_29960768 0.0 29960768 G T 22
23 22 snp_22_30253157 0.0 30253157 A G 23
24 22 indel:4D_22_30663957 0.0 30663957 G GCAGA 24
25 22 snp_22_30901592 0.0 30901592 C T 25
26 22 snp_22_30937512 0.0 30937512 G A 26
27 22 snp_22_31024375 0.0 31024375 A C 27
28 22 snp_22_31102820 0.0 31102820 G A 28
29 22 snp_22_31496200 0.0 31496200 T C 29
.. ... ... ... ... ... ... ..
70 22 snp_22_43779140 0.0 43779140 T C 70
71 22 indel:1D_22_43820821 0.0 43820821 C CG 71
72 22 snp_22_44052552 0.0 44052552 C T 72
73 22 snp_22_44162123 0.0 44162123 A G 73
74 22 snp_22_44657401 0.0 44657401 A G 74
75 22 snp_22_44933193 0.0 44933193 C A 75
76 22 snp_22_45136558 0.0 45136558 G A 76
77 22 snp_22_45442509 0.0 45442509 C A 77
78 22 snp_22_46289699 0.0 46289699 C T 78
79 22 snp_22_46650858 0.0 46650858 C A 79
80 22 snp_22_46665209 0.0 46665209 A G 80
81 22 snp_22_46870068 0.0 46870068 T C 81
82 22 snp_22_46938676 0.0 46938676 C T 82
83 22 snp_22_47061834 0.0 47061834 A G 83
84 22 snp_22_47500904 0.0 47500904 T C 84
85 22 snp_22_47586093 0.0 47586093 C T 85
86 22 snp_22_47627719 0.0 47627719 T C 86
87 22 snp_22_47772918 0.0 47772918 C G 87
88 22 indel:3I_22_48207120 0.0 48207120 CCAG C 88
89 22 snp_22_48439843 0.0 48439843 C A 89
90 22 snp_22_48740730 0.0 48740730 T C 90
91 22 indel:16D_22_48777234 0.0 48777234 A AACCCAGGAGAGGATCG 91
92 22 snp_22_48836042 0.0 48836042 G A 92
93 22 snp_22_49010580 0.0 49010580 T C 93
94 22 snp_22_49335866 0.0 49335866 A G 94
95 22 indel:4D_22_49340059 0.0 49340059 G GAGAC 95
96 22 snp_22_49362308 0.0 49362308 C T 96
97 22 snp_22_49473688 0.0 49473688 T C 97
98 22 snp_22_49568955 0.0 49568955 G A 98
99 22 snp_22_50837415 0.0 50837415 A G 99
[100 rows x 7 columns]
------------------- Genotype -------------------
fid iid father mother gender trait i
0 0 HG00105 0 0 0 -9 0
1 0 HG00107 0 0 0 -9 1
2 0 HG00115 0 0 0 -9 2
3 0 HG00132 0 0 0 -9 3
4 0 HG00145 0 0 0 -9 4
5 0 HG00157 0 0 0 -9 5
6 0 HG00181 0 0 0 -9 6
7 0 HG00308 0 0 0 -9 7
8 0 HG00365 0 0 0 -9 8
9 0 HG00371 0 0 0 -9 9
10 0 HG00379 0 0 0 -9 10
11 0 HG00380 0 0 0 -9 11
12 0 HG01789 0 0 0 -9 12
13 0 HG01790 0 0 0 -9 13
14 0 HG01791 0 0 0 -9 14
15 0 HG02215 0 0 0 -9 15
16 0 NA06985 0 0 0 -9 16
17 0 NA07346 0 0 0 -9 17
18 0 NA11832 0 0 0 -9 18
19 0 NA11840 0 0 0 -9 19
20 0 NA11881 0 0 0 -9 20
21 0 NA11918 0 0 0 -9 21
22 0 NA12005 0 0 0 -9 22
23 0 NA12156 0 0 0 -9 23
24 0 NA12234 0 0 0 -9 24
25 0 NA12760 0 0 0 -9 25
26 0 NA12762 0 0 0 -9 26
27 0 NA12776 0 0 0 -9 27
28 0 NA12813 0 0 0 -9 28
29 0 NA18488 0 0 0 -9 29
.. .. ... ... ... ... ... ...
435 0 NA20785 0 0 0 -9 435
436 0 NA20786 0 0 0 -9 436
437 0 NA20787 0 0 0 -9 437
438 0 NA20790 0 0 0 -9 438
439 0 NA20792 0 0 0 -9 439
440 0 NA20795 0 0 0 -9 440
441 0 NA20796 0 0 0 -9 441
442 0 NA20797 0 0 0 -9 442
443 0 NA20798 0 0 0 -9 443
444 0 NA20799 0 0 0 -9 444
445 0 NA20800 0 0 0 -9 445
446 0 NA20801 0 0 0 -9 446
447 0 NA20802 0 0 0 -9 447
448 0 NA20803 0 0 0 -9 448
449 0 NA20804 0 0 0 -9 449
450 0 NA20805 0 0 0 -9 450
451 0 NA20806 0 0 0 -9 451
452 0 NA20807 0 0 0 -9 452
453 0 NA20808 0 0 0 -9 453
454 0 NA20809 0 0 0 -9 454
455 0 NA20810 0 0 0 -9 455
456 0 NA20811 0 0 0 -9 456
457 0 NA20812 0 0 0 -9 457
458 0 NA20813 0 0 0 -9 458
459 0 NA20814 0 0 0 -9 459
460 0 NA20815 0 0 0 -9 460
461 0 NA20816 0 0 0 -9 461
462 0 NA20819 0 0 0 -9 462
463 0 NA20826 0 0 0 -9 463
464 0 NA20828 0 0 0 -9 464
[465 rows x 7 columns]
BIMBAM file formats¶
Phenotype:
$ limix see phenotype.gemma --filetype=bimbam-pheno
0 1 2
0 1.2 -0.3 -1.5
1 NaN 1.5 0.3
2 2.7 1.1 NaN
3 -0.2 -0.7 0.8
4 3.3 2.4 2.1
[5 rows x 3 columns]
HDF5¶
The following command shows the hierarchy of a HDF5 file:
$ limix see small_example.hdf5
/
+--genotype
+--col_header
| +--chrom [|S8, (100,), None]
| +--pos [int64, (100,), None]
+--matrix [uint8, (183, 100), None]
+--row_header
+--sample_ID [|S7, (183,), None]
CSV¶
CSV files have their delimiter automatically detected and a preview can be shown as
$ limix see small_example.csv
Reading small_example.csv... done (0.08 seconds).
0 1 2 3 4 5 6 ... 459 460 461 462 463 464 465
0 snp_22_16050408 A A A A A A ... B B B B B B B
1 snp_22_16050612 A A A A A A ... B B B B B B B
2 snp_22_16050678 A A A A A A ... B B B B B B B
3 snp_22_16051107 A A A A A A ... B B B B B B B
4 snp_22_16051249 A A A A A A ... B B B C C B B
[5 rows x 466 columns]

{kind=link}
{kind=link}
GWAS¶
$ limix scan --help
Usage: limix scan [OPTIONS] PHENOTYPES_FILE GENOTYPE_FILE
Perform genome-wide association scan.
This analysis requires minimally the specification of one phenotype
(PHENOTYPES_FILE) and genotype data (GENOTYPE_FILE).
The --filter option allows for selecting a subset of the original dataset
for the analysis. For example,
--filter="genotype: (chrom == '3') & (pos > 100) & (pos < 200)"
states that only loci of chromosome 3 having a position inside the range
(100, 200) will be considered. The --filter option can be used multiple
times in the same call. In general, --filter accepts a string of the form
<DATA-TYPE>: <BOOL-EXPR>
where <DATA-TYPE> can be phenotype, genotype, or covariate. <BOOL-EXPR> is
a boolean expression involving row or column names. Please, consult
`pandas.DataFrame.query` function from Pandas package for further
information.
Options:
--covariates-file TEXT Specify the file path to a file containing the
covariates.
--kinship-file TEXT Specify the file path to a file containing the
kinship matrix.
--lik TEXT Specify the type of likelihood that will described
the residual error distribution.
--filter TEXT Filtering expression to select which phenotype,
genotype loci, and covariates to use in the
analysis.
--output-dir TEXT Specify the output directory path.
-h, --help Show this message and exit.